| A ribosome doing its thing. From Wikipedia |
In that same article I mentioned my plans to use a high tech, and yet strangely cheaper, method. This method will be outlined in this post. This method uses a technique called "polymerase chain reaction", a method used to copy specific DNA sequences to identify yeast. We amplify a specific portion of the yeast (or bacterial) genome and then sequence the amplified DNA. The resulting sequence is then compared to a DNA database to ID the yeast/bacteria in the sample. Despite the high tech nature of the method, it is relatively cheap. Low-cost PCR kits, combined with crude DNA isolations and low-cost sequencing keep prices down to roughly $10/strain. While you wouldn't want to screen hundreds of strains, this method is more than affordable to identify the final candidates in the wild yeast project.
All the good stuff is below the fold...
Legend
- Early Publication Note
- Brief Outline of the Method
- WTFerment is PCR?
- WTFerment is a Ribosome & why do we use it?
- Protocol: Crude DNA isolation
- Protocol: ITS PCR
- Protocol: Using BLAST to Analyze the DNA sequence
- Resources
Early Publication Note:
Due to repeated emails (seriously guys/gals, its OK to post comments) I am posting this before running some actual tests. Hopefully the real-world sequencing tests will work out well. I hope to have the real-world examples completed in the next few weeks.
Brief Outline of the Method
Before going into the nitty-gritty I think its worth briefly explaining how this all works, in a way that is free of all the technical terminology. If you find some of the terminology confusing it may be worth going back and reading the Taxonomy section of my first post on identifying yeasts.
This method is simple in concept, if not in implementation. The end-goal is to sequence a small part of the yeast's genome, and to use that sequence to then identify the yeast. Not any sequence will do - we need something conserved enough between different types of yeast/bacteria so that PCR can amplify it, but we also need there to be enough differences so that we can identify different species (genera, families, etc). The best part of the genome for this is the part that encodes ribosome's - for reasons outlined below.
To make this happen we start by growing up a bit of yeast (or bacteria), and then crudely purify the DNA. We need only a crude purification, as the next step is to use a technology termed PCR to amplify the part of the ribosome that we will use for sequencing. We then send off the PCR product for sequencing. Finally, we take the sequence and use it to search a DNA sequence database, hopefully matching it up with a known yeast/bacterial genome. Vola - we've identified the yeast.
This method is simple in concept, if not in implementation. The end-goal is to sequence a small part of the yeast's genome, and to use that sequence to then identify the yeast. Not any sequence will do - we need something conserved enough between different types of yeast/bacteria so that PCR can amplify it, but we also need there to be enough differences so that we can identify different species (genera, families, etc). The best part of the genome for this is the part that encodes ribosome's - for reasons outlined below.
To make this happen we start by growing up a bit of yeast (or bacteria), and then crudely purify the DNA. We need only a crude purification, as the next step is to use a technology termed PCR to amplify the part of the ribosome that we will use for sequencing. We then send off the PCR product for sequencing. Finally, we take the sequence and use it to search a DNA sequence database, hopefully matching it up with a known yeast/bacterial genome. Vola - we've identified the yeast.
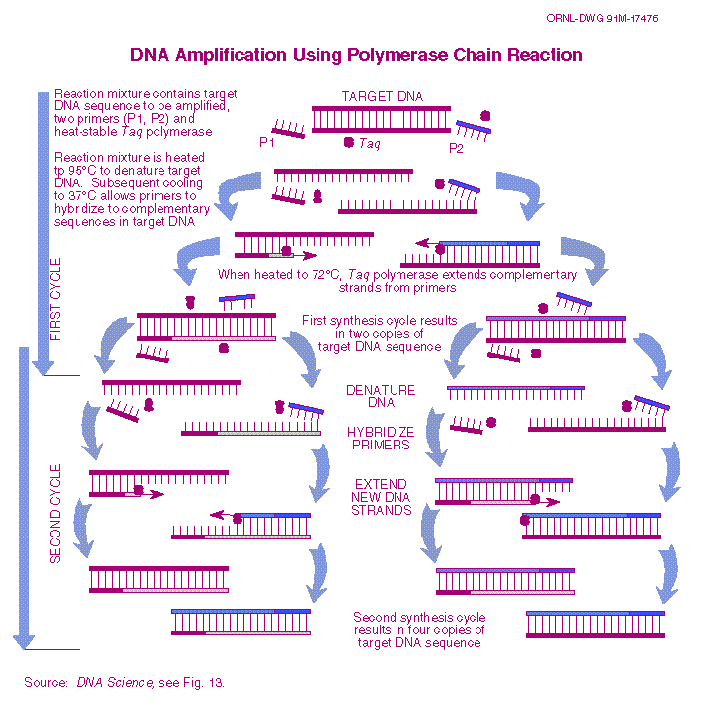
WTFerment is PCR?
So at this point you may be asking "that's nice, but what the ferment is PCR". PCR is short-form for "polymerase chain reaction"; a longer way of saying "DNA photocopier". But unlike a photocopier, we can very specifically amplify (copy) exact portions of the genome. This selective amplification ability allows us to take a very small amount of crudely purified DNA and then amplify out a large quantity of the desired sequence.So how does it work?
PCR starts by heating the DNA to just below the boiling point of water (usually 96C). This causes the DNA's normally double stranded structure to 'unwind' into single strands - a process we refer to as 'DNA melting' or 'denaturation'. In the same solution as our melting DNA are primers - short sequences of single-stranded DNA that match up with the ends of the portion of the yeast/bacterial genome we wish to amplify. We then cool the sample to between 45C and 60C; this causes the DNA to try and become double-stranded again ('annealing'). However, we setup our initial conditions such that there is a lot of primers and not a lot of genomic DNA. As such most of the double-stranded DNA we get is regions where our primers are bound to the ends of the DNA sequence we are trying to amplify. We then warm the sample slightly - usually to round 72C - which activates a DNA-copying enzyme that then extends the primers such that we copy the DNA between the primers. The process is then repeated, with every repeat doubling the number of copies of the desired DNA sequence.
 |
| Graphical Description of PCR Click for Full-Sized From: http://www.ucl.ac.uk/~ucbhjow/b200/pcr.htm |
WTFerment is a Ribosome & why do we use it?
Hopefully that made sense, but if you forgot your highschool biology (as most of us tend to do), you're now asking "what the ferment is a ribosome", and perhaps "why a ribosome and not something else".
So why target ribosomes? Ribosomes themselves are a bit unusual, and are made of a mix of RNA and protein. The RNA portion is so very important to the functioning of the ribosome that most mutations lethally damage the ribosome. As such, ribosomes evolve very slowly - except for one part. During their production, all of the RNA part of a ribosome is copied from the genomic DNA as one intact strand. This strand is then cut into the 4 separate pieces of RNA that comprise the final ribosome. Between two of these pieces is a short segment that is not part of the ribosome termed the "internal transcribed spacer" (ITS) region. Since this piece is simply cut out, and mutations do not harm it, it evolves quite quickly.
 |
| Yeast ribosome gene structure. From: http://biology.duke.edu/fungi/mycolab/ |
So we are targeting a very specific part of the ribosome. Specifically, we are using primers that bind to the end of the 18S ribosomal subunit and to the start of 5.8S ribosomal subunit, thus amplifying the region between them (i.e. the ITS). These structures can be seen in the figure to the right; the leftmost thick black bar is the 18S, the next (very short) black bar is the 5.8S. The ITS is the gap between them. By amplifying - and then sequencing - the ITS, we can figure out what species of yeast we have.
There are a number of methods to isolate yeast DNA. The method outlined here is a cruder form of the high-quality DNA isolation found in this protocol. Additional protocols can be found here.
| Step 1: Grow up a 1ml culture of the desired yeast in YPD or 1.020 wort. | |||
| Step 2: Centrifuge 5 minutes at 20,000 x G to pellet the yeast. | ||
Harji Buffer:
| Step 3:
|
||
Step 4: Repeat 3 times:
|
|||
| Step 5: Separate Proteins By:
|
||
Step 6: Separate the DNA:
|
|||
Step 7: Almost there!
|
|||
Step 8: Done!
|


{kind=link}
These protocols are based on those described in Brewhouse-Resident Microbiota Are Responsible for Multi-Stage Fermentation of American Coolship Ale.
First, the appropriate primers need to be ordered. Two are needed for yeast, and two different ones for bacteria. Two are needed for each, since a different primer binds to either side of the ITS region. The ribosomes are different enough between bacteria & yeast that we cannot use the same primers (note: the 5' and 3' demarcates the direction of the DNA strand). The bacterial primers don't technically amplify an ITS region, but the principal is the same - our primers bind conserved regions, and amplify a section that varies between species.
ITS Primers For Yeast:
First, the appropriate primers need to be ordered. Two are needed for yeast, and two different ones for bacteria. Two are needed for each, since a different primer binds to either side of the ITS region. The ribosomes are different enough between bacteria & yeast that we cannot use the same primers (note: the 5' and 3' demarcates the direction of the DNA strand). The bacterial primers don't technically amplify an ITS region, but the principal is the same - our primers bind conserved regions, and amplify a section that varies between species.
ITS Primers For Yeast:
ITS1: 5′ - TCCGTAGGTGAACCTGCGG -3'
ITS4: 5′ - TCCTCCGCTTATTGATATGC -3'
ITS Primers For Bacteria:
Uni331F 5′- TCCTACGGGAGGCAGCAGT -3′
1492R 5′- GGTTACCTTGTTACGACTT -3′
To begin: Mix the purified DNA, primers, and reagents that come in your PCR kits, as per manufacturers instructions Generally, you want to use 0.5ul to 1ul of the purified DNA in a 30ul to 50ul PCR reaction. Then amplify the DNA using the following amplification protocol:
- Activate your PCR enzyme as per manufacturers instruction; this is usually an initial heat for 2-3min at 95C.
- Heat to 95C for 30-60 seconds to melt the DNA.
- Let the primers bind the DNA by cooling to 50C (yeast) or 66C (bacteria) for 30-60 seconds.
- Warm to the active temperature of your PCR enzyme; usually 72C. Hold at this temperature for long enough to amplify 1000bp of DNA (usually 30-60 seconds)
- Repeat steps 2-4 44 more times.
- Perform a final extension, 72C for 5-10 minutes.
Check/Purify DNA Sample:The next step is to use a DNA gel to check that the PCR worked, and to purify the DNA. There are so many methods to do this that I cannot mention them all here. In short form, run out the PCR reaction on a 2% agarose gel. Post-stain with ethidium bromide and image on a UV imager. If PCR is successful, cut out the band(s) (sometimes there will be 2) corresponding to the amplified region (usually 400 to 1000 basepairs in size) and purify into 12ul water with a glassmilk kit (i.e. geneclean II kit).
Sequencing:Sequencing is done using contract facilities. Mine charges $5 per sequence. Samples need to be prepared as per the facilities instructions.
Sequencing:Sequencing is done using contract facilities. Mine charges $5 per sequence. Samples need to be prepared as per the facilities instructions.
Protocol: Using BLAST to Analyze the DNA Sequence
 |
| BLAST analysis of the published Saccharomyces cerevisiae ITS region |
We now have a sequence of our yeast's ITS region - so what do we do with it? The answer is we search a DNA database for similar sequences using Basic Local Alignment Search Tool (BLAST). There are a number of BLAST engines out there, but for this project two come to mind. The NCBI runs the largest database of DNA sequences and has an excellent BLAST server (link). The Yeast Genome Database is more focused on fungal genomes, but has a less intuitive BLAST interface. Searching either should give us a usable result.
In the picture to the right I show the results of blasting the published Saccharomyces cerevisiae ITS region. The red indicates highly accurate matches - in all cases, clicking on the red bar will take you to additional information. In all of these cases, the match is to a strain of Saccharomyces cerevisiae.
In the picture to the right I show the results of blasting the published Saccharomyces cerevisiae ITS region. The red indicates highly accurate matches - in all cases, clicking on the red bar will take you to additional information. In all of these cases, the match is to a strain of Saccharomyces cerevisiae.
To try this yourself:
- Browse to NCBI Blast, then click the "Nucleotide" link half-way down the page. This will take you to a search engine for DNA sequences.
- Copy your DNA sequence file into the window titled 'Enter accession number(s), gi(s), or FASTA sequence(s)'. As an example, copy the "defult" Saccharomyces cerevisiae ITS DNA sequence from this link.
- Ignore the rest of the options on the BLAST page, click the blue "Blast" button.
- After a few seconds, you will get your list of matched sequences.
cggtcttgctaggcttgtaagtgcctttcttgctattccaaacgatgagcgatttcggtgctctttttatgggacaattcaaacagtttcaatacaatacactgtggaggtttcatatctttgcaactttttctttgggcattcgagcaatcgggggctagacgtaacaaacacaaaccattttatctattcaaaaaatttttgtcaaaaacaagaactttcgcaacgggaaattttaaaacattcaaaactttcaacaaaggatctcttggttctcgcgtcgtagaagaacgcagcgaagtgcgatacgtaatgtgaattttataattccgtgaatcatcgaatctttgaacgcacattgcgccccttagtattccaggggcatgcctgttcgagcgccaattccttctcagacattctgtatggtagtgagtgatactctttggatttaacttgaaattgatggccttttcattggaagttttgtttccaaaaagaggtttct
When BLASTed you'll get an image almost the same as above (some of the bars may be different colours or partial in length), but looking at the table below the image you will see a table with the following critical information:
 |
| Data Table - click to zoom in. |
The key numbers are the 'Query Cover' and 'Max Ident' columns. "Query Cover" indicates the percentage of the sequence we entered into the BLAST algorithm (the query) that matched to a sequence in the database. 100% means everything we entered matched up. Sometimes only part of the sequence will match, and this number will drop. The 'Max Ident' column indicates how well the query sequence that overlapped with the database sequence matched the database exactly. So a 50% 'Query Cover' with 99% 'Max Ident' means that 50% of the sequence we entered matched almost-perfectly with the database. Realistically, our 'Query Cover' will be close to 100% and our 'Max Ident' will be somewhere between 95% and 100%.
So which one is our yeast? Its probably the one at the top of the list, which is sorted by a score which incorporates both the 'Query Cover' and 'Max Ident' values.
So which one is our yeast? Its probably the one at the top of the list, which is sorted by a score which incorporates both the 'Query Cover' and 'Max Ident' values.
Resources:
- PCR Primers That Amplify Fungal rRNA Genes from Environmental Samples - Free scientific article outlining a method to ID fungi (including yeast) based on their ribosomal sequences.
- Conserved primer sequences for PCR amplification and sequencing from nuclear ribosomal RNA - webpage outlining primers and methods to ID yeast by sequencing.
- Brewhouse-Resident Microbiota Are Responsible for Multi-Stage Fermentation of American Coolship Ale - Free scientific article on the yeasts/bacteria found in lambic-style beer & the use of sequencing primers to ID the species within the sample.
- List of yeast protocols, including the DNA isolation method mentioned here.
- NCBI BLAST - search multiple DNA databases for genome sequences.
- Yeast Genome Database - database of yeast & other fungi genomes, includes a BLAST feature.
Hi,
ReplyDeletethanks for this great tutorial. I have a question,
how to prepare a Harju buffer using the info in your post? Just mix the reagents and top it off with water?
Robert,
ReplyDeleteThat is exactly how it is done. It is easiest to make a 10X tris, 100x EDTA and 20% SDS solution ahead of time. These are then diluted, along with the triton and NaCl into water. For example, for 10ml:
To 8.2ml of water add:
-1ml of 10X tris (100mM @ pH 8)
-100ul of 100x EDTA (100mM)
-500ul of 20% SDS
-200ul of triton X100
-58mg of NaCl
That said, the DNA isolation method worked imperfectly (see the followup posts #1 and #2). I am working on a simpler/cheaper method that may work better - its described at the end of followup #2, and I should be posting my first results using it in about a month.
Hi Bryan,
Deletetoday i tried both methods of DNA isolation (ligase buffer [LB] and boil/freeze [BF]), both worked fine. The yield from the LB was several magnitudes greater then the BF method.
Sample 1 LB DNA 663ng/µl
Sample 2 LB DNA 1872.3ng/µl
Sample 1 BF DNA 1.2ng/µl
Sample 2 BF DNA 2.3ng/µl
Which might also explain your unsatisfatory PCR results with the buffer. I had to dilute the LB samples to around 100ng/µl to successfully perform a PCR.
Robert
Robert,
DeleteIn my case I'm confident DNA concentration was not an issue - I spec'd the DNA and found that there was a heavy protein contamination (plus an undefinable, but modest, amount of DNA) in the samples that didn't work. The ones that worked had little protein contamination. I suspect I over-loaded the two samples that didn't work, and saturated the system.
Bryan
Hi there, what PCR machine and PCR kit did you use?
ReplyDeleteThanks,
Alex
We make our own Taq polymerase, and the PCR's were run on an old eppidorff thermocycler.
DeleteHi, how specific are those ITS primers for different species of yeast and bacteria, i.e. will they work with things like Brettanomyces, Lactobacillus, or Pediococcus, or just common brewing yeasts like Saccharomyces cerevisiae? Thanks!
ReplyDeleteITS are specific for eukaryotas or prokariotes - i.e. the primers listed for 'yeast', above, will work for all fungi (including sacc and brett) as well as any other eukariote (e.g. plants, people, purple starfish). The bacterial primers listed above will work for all bacteria including lactobacillus and pediococcus, but will not work for eukariotes.
DeleteFor lacto/pedio (and all other bacteria) you